




Multipronged Management of Interstitial Lung Disease

Dr. Fernando J. Martinez
More than 200 distinct pulmonary disorders fall under the umbrella of interstitial lung disease (ILD). These are complex diseases that often lead to progressive scarring of the lungs and have a prognosis worse than many cancers. Pulmonologists at NewYork-Presbyterian/Weill Cornell Medical Center are developing a new paradigm to confront idiopathic pulmonary fibrosis (IPF) and other forms of progressive fibrotic ILDs, including chronic hypersensitivity pneumonitis (CHP).
“We have a robust program for evaluating patients, assigning diagnosis – which is very challenging in these disorders – providing available therapies, and initiating multiple therapeutic strategies,” says Fernando J. Martinez, MD, MS, Chief, Division of Pulmonary and Critical Care Medicine at NewYork-Presbyterian/Weill Cornell. Dr. Martinez is a premier translational researcher in chronic lung disease and is internationally recognized for his seminal studies in the phenotypic classification and clinical interventions in interstitial lung disease.
Importance of Correct Diagnosis
According to Dr. Martinez, ILD takes on a variety of forms and it is often difficult to understand if a patient has interstitial lung disease and what caused it. “To arrive at an accurate diagnosis requires taking a detailed history, performing a thorough physical exam, ordering the appropriate testing, including a high resolution chest CT, and reviewing imaging and biopsy results, if performed, with a team of specialists in ILD,” he says. “We work closely as part of a multidisciplinary team of pulmonologists, pathologists, and radiologists who meet bimonthly to review cases and make sure patients are getting the right diagnosis and the right treatment.”
“The Weill Cornell interstitial lung team encompasses many different subspecialties and allows us to apply our expertise at a very high level,” notes Robert J. Kaner, MD, a Weill Cornell pulmonologist and a leading expert in interstitial lung diseases. “In our testing, we’re looking for patterns of abnormality that would suggest that the gas exchanging part of the lung is being affected. Radiologists use terms like ground glass opacity, reticulation, architectural distortion, traction bronchiectasis, and honeycombing to describe some of the typical findings of ILD. The ones that indicate scarring are reticulation, traction bronchiectasis, and honeycombing. When we see those findings, we know that scarring is occurring in the lung and not just inflammation.”

Dr. Robert J. Kaner
Dr. Kaner describes earlier trials that have changed the standard of care for treating idiopathic pulmonary fibrosis, which affects one in 200 adults over the age of 65 in the U.S. “The very first randomized placebo control trial for IPF, a study of beta interferon, turned out to be a negative study, but it proved for the first time that you could conduct a multicenter study in this rare disease and achieve a valuable result,” he says. “After that study, the National Heart, Lung and Blood Institute was convinced that they should fund a clinical research network to carry out multicenter trials for drug treatments for IPF. The Idiopathic Pulmonary Fibrosis Clinical Research Network was established with 11 sites in the U.S., including Weill Cornell where I served as principal investigator.”
The IPF Network study tested the efficacy of immunosuppressive therapy in IPF, the standard of care at the time. The three-drug regimen of azathioprine, prednisone, and N-acetylcysteine not only failed to show benefit in IPF, but the trial was stopped early because of harm in the group that took the three-drug combination. Those early trials set the groundwork that led to the first FDA-approved anti-fibrotic treatments for IPF.
“That was a very important observation,” says Dr. Kaner. “Nintedanib and pirfenidone are anti-fibrotic drugs and are not immunosuppressive. While they are not curative, they do slow down the progression. These data, taken together, inform our approach to ILD, where we spend a lot of time and effort to make sure that we have the correct diagnosis.”
“At this point, it’s not known whether reversal of ILD is possible, though there are some smaller phase 2 studies of new drugs that suggest that that might be the case, particularly for people in the earlier stages of the disease,” continues Dr. Kaner. “Our goal for most patients is to stabilize the abnormality. In people who have the more inflammatory types of ILD, we can help to improve their radiographic findings and symptoms with immunosuppressive medication. In the diseases that are characterized purely by scarring in the lung, our hope is to stabilize the scarring or slow down its progression.”
A Comprehensive Research Program
NewYork-Presbyterian/Weill Cornell serves as the primary site for a number of ongoing research studies aimed at improving understanding of ILD and developing better treatments and participates in several national and global clinical trials and registries.
Applying Precision Medicine to IPF
In 2019, Dr. Martinez was appointed a co-principal investigator of the PRECISIONS (Prospective tReatment EffiCacy in IPF uSIng genOtype for Nac Selection), a global multicenter study and the first clinical trial to apply the principles of precision medicine to the treatment of patients with idiopathic pulmonary fibrosis. Funded by the NIH, PRECISIONS aims to determine if N-Acetyl-cysteine (NAC) is an effective treatment in a subset of IPF patients who carry a particular genetic variant and to define genetic and molecular signatures to distinguish IPF from non-IPF interstitial lung disease.
“The PRECISIONS enterprise is taking diagnosis and treatment for lung disease to a new level in personalized medicine,” says Dr. Martinez. “Such targeted therapy may be a significant advance for a specific group of patients, providing new insights about the biology of the disease that may drive the field into a new era.”
PRECISIONS incorporates multiple components, including performing the first truly precision-based trial in patients with IPF to study a relatively easily available therapy that perhaps may prevent death in a fraction of patients that have a very specific genetic makeup. “We’ve learned from the cancer investigators – this is their paradigm,” notes Dr. Martinez. “We’re also completing a series of very detailed assessments of molecular factors that can segregate patients to different groups and how those groups could be used for diagnosis and therapy.”
Participants in the NAC trial are being recruited through the Pulmonary Fibrosis Foundation’s Patient Registry and Biorepository. NewYork-Presbyterian/Weill Cornell is one of 42 collaborating care centers nationwide that have enrolled patients in the registry.
Identifying ILD in its Earliest Stages

Dr. Anna J. Podolanczuk
Among other investigations underway are those of Weill Cornell pulmonologist Anna J. Podolanczuk, MD, MS, an NIH-funded investigator studying novel risk factors for pulmonary fibrosis. Her research focuses on understanding early or subclinical interstitial lung disease, characterizing imaging and blood biomarkers of pulmonary fibrosis, the role of environmental exposures, and the connection between cardiovascular disease and pulmonary fibrosis.
“The best way to understand why people get pulmonary fibrosis is to study early disease in humans,” says Dr. Podolanczuk, whose work focuses on identifying individuals at the earliest stages of ILD and to conduct treatment studies before the disease becomes symptomatic. “We do that primarily by studying the progression of incidentally detected interstitial lung abnormalities, also called ILA, on CT scans done for other reasons. I collaborate with our expert thoracic radiologist to identify patients with early ILD who have undergone CT scans for lung cancer screening. We’ve also worked with large cohorts of patients who have been enrolled in cardiovascular disease studies and have repurposed their CT scans to look at the lungs.”
Dr. Podolanczuk investigates risk factors for early disease, both by visually assessing CT scans for interstitial changes, as well as using advanced computer-generated measures to detect very subtle changes. “We then try to identify new blood biomarkers that might be associated with these early interstitial changes,” she says. “We’re looking at high-density lipoproteins and the components of HDL, specifically apolipoprotein A1, as a protective factor and future therapeutic target. I’m also interested in environmental exposures and other acquired factors, and how they relate to the development and progression of pulmonary fibrosis.”
Dr. Podolanczuk is encouraged by the number of trials and potential treatments for pulmonary fibrosis in the pipeline. “I tell my patients that there’s so much hope now,” she says. “For example, a landmark study came out last year that showed nintedanib not only works for IPF, but also for patients with other kinds of progressive pulmonary fibrosis. That’s something that we’ve all been waiting for. Now I can offer patients options for disease-modifying therapy to help them live longer lives.”
Role of Lung Lymphatics

Dr. Hasina Outtz Reed
Hasina Outtz Reed, MD, PhD, a physician-scientist who is trained in Pulmonary and Critical Care Medicine, as well as Cellular and Molecular Biology, is studying the role that lymphatic impairment plays in lung disease and seeks to uncover a link between lymphatic dysfunction and lung injury. “The fundamental question of my research program is how do the lung lymphatics work and how do they contribute to the response to lung injury and lung disease,” says Dr. Outtz Reed. “Using genetic mouse models, high-level imaging, and other molecular tools, we’re able to investigate the lymphatic vasculature and ask these questions in ways that hopefully will have important consequences for understanding human disease.”
As Dr. Outtz Reed explains, markers used to identify the lymphatics in the lungs are different than markers used to distinguish the lymphatics in other organs in the body. “Lymphatics in the lung not only drain proteins and fluid from the lungs, but they also traffic immune cells,” she says. “There is a lot of interplay between the lung and the lymphatics that drain them. And the crosstalk between these vessels and the immune system and lung function is what we’re focused on.”
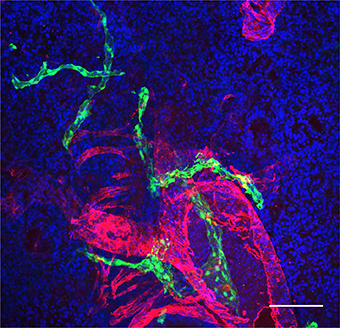
Mouse lung lymphatics (green), in close proximity to an airways and pulmonary artery (red) (Courtesy of Dr. Hasina Outtz Reed)
“It was presumed that the major role of lymphatics is to primarily drain fluid from the lung tissue, but it turns out that’s not the case,” continues Dr. Outtz Reed. “We’ve learned that they play an integral role in the response to inflammation and their function in immune cell trafficking. Some of our early discoveries showed the extent to which lymphatic dysfunction in the lungs can lead to changes in the inflammatory and immune cell milieu that has very important consequences for lung injury. This has broad reaching consequences, whether it be for pulmonary fibrosis, emphysema, and even lung transplant. The lung lymphatics as a therapeutic target in these diseases has been entirely unexplored, and it is exciting to think that this research may lead to discoveries that make a difference for diseases for which there are not a lot of therapies.”
Dr. Outtz Reed and her colleagues at Memorial Sloan Kettering Cancer Center are also exploring the development of nanoparticle-based therapeutics that target the lung lymphatics. “We have ongoing studies of lung fibrosis models in mice,” she says. “In the future, we will test whether we can affect these processes using novel therapeutics and whether we can encapsulate lymphatic-targeted drugs into nanoparticles.”
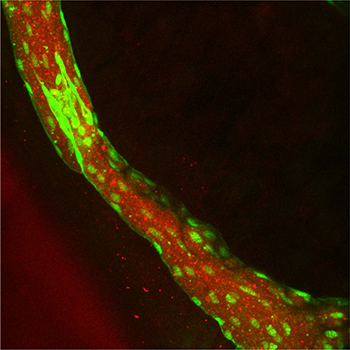
Rhodamine (red)-labelled nanoparticles delivered to a mouse lung lymphatic (green)
“These mechanistic and basic questions that we’re asking are very exciting and haven’t really been asked before,” adds Dr. Outtz Reed. “I think we’re going to be able to make a lot of breakthroughs in how we think about lung diseases in completely different ways.”
Elevating Outcomes Measures for CHP

Dr. Kerri I. Aronson
Kerri I. Aronson, MD, MS, a critical care medicine specialist at Weill Cornell, has a particular clinical interest and research focus on chronic hypersensitivity pneumonitis, including patient-centered outcomes and the challenges associated with antigen identification and avoidance. The disease is particularly challenging to manage as there are no universally accepted diagnostic criteria, few FDA-approved therapies, and no standardized approach to identifying an antigen. CHP presents with a variety of symptoms that impact patient function and overall well-being.
“Patients with CHP have variable clinical courses, and the tests we currently use to measure disease status, such as pulmonary function testing, are not the outcomes that are truly important to patients,” says Dr. Aronson. “The impact that CHP has on patients’ quality of life can be profound. As more CHP patients are enrolled in therapeutic trials, there is a need for a patient-centered outcome specific to CHP patients.”
Dr. Aronson had previously identified unique factors that influence quality of life from the perspective of patients. These include lack of knowledge, uncertainty, impact on self-perception and identity, and the complex psychosocial problems associated with antigen identification and avoidance.
To address the need for outcome measures that better represent patient concerns, Dr. Aronson and her colleagues in the Division of Pulmonary and Critical Care Medicine, General Internal Medicine, and Genetic Medicine at Weill Cornell recently established and documented content validity of a disease-specific health-related quality-of-life survey (HRQOL) for CHP patients. Their process and results are reported in detail in the January 14, 2021 Journal of Patient-Reported Outcomes. The study was the first to describe the documentation of content validity for a disease-specific HRQOL instrument for CHP patients and it is now ready for psychometric testing in further validation studies.
“Our long-term goal is to develop a reliable approach to assess and quantify therapeutic effects that are most important to patients living with CHP,” notes Dr. Aronson.

Related Publications
